Gruppe 1
Pulmonale arterielle Hypertonie (PAH)
Definition
Die pulmonale arterielle Hypertonie (PAH) bildet gemäß WHO- und Nizza-Klassifikation die Gruppe 1 der pulmonalen Hypertonie (PH).1–3 Bei der PAH liegt eine präkapilläre PH ohne andere erkennbare Ursachen vor – im Unterschied zur präkapillären PH infolge chronischer Thromboembolien (CTEPH) oder Lungenerkrankungen.1
Hämodynamische Definition der PH-Gruppe 1 (PAH präkapilläre PH):1 | |
|---|---|
Mittlerer Pulmonalarteriendruck (mPAP) | > 20 mmHg |
Pulmonalarterienverschlussdruck (PAWP) | ≤ 15 mmHg |
Pulmonalvaskulärer Widerstand (PVR) | > 2 WU (Wood Unit) |
Die PAH ist eine besonders schwerwiegende Form der PH.1,4 Diagnostik und Therapie sollten in enger Zusammenarbeit zwischen spezialisierten PH-Expertenzentren und der/dem niedergelassenen Ärztin oder Arzt erfolgen.1
Klassifikation
Die Pathophysiologie der PAH ist multifaktoriell.1 In der aktuellen WHO-Klassifikation wird die PAH untergliedert in:1,3,4
Idiopathische PAH (IPAH)
Nicht-Responder in der Vasoreaktivitätstestung
Akute Responder in der Vasoreaktivitätstestung
Hereditäre PAH (HPAH)
Medikamenten- oder Toxin-induzierte PAH (DPAH)
Der Zusammenhang zwischen der Exposition gegenüber Medikamenten oder Toxinen und PAH wird wie in der Tabelle eingestuft:
Definite association | Possible association |
|---|---|
Aminorex Benfluorex Dasatinib Dexfenfluramine Fenfluramine Methamphetamines Toxic rapeseed oil | Alkylating agents (cyclophosphamide, mitomycin C)a Cocaine |
Tab. 1: Arzneimittel und Toxine, welche definitiv oder möglicherweise mit PAH assoziiert werden. Modifiziert nach 1.
PAH assoziiert mit:
Bindegewebserkrankungen (CTD)
HIV-Infektion
Portaler Hypertension (PoPH)
Angeborenen Herzfehlern (CHD)
Schistosomiasis
PAH mit offensichtlichen Hinweisen auf eine venöse/kapilläre Beteiligung (PVOD: pulmonale veno-okklusive Erkrankung, PCH: pulmonale kapilläre Hämangiomatose)
Persistierende PH des Neugeborenen (PPHN)
Epidemiologie und Risikofaktoren
Die PAH ist eine seltene Erkrankung („Orphan Disease“):1
Inzidenz: ca. 6 Fälle pro 1 Mio. Erwachsene
Prävalenz: 48–55 Fälle pro 1 Mio. Erwachsene
Häufigkeit in PAH-Registern: IPAH (50–60 %) > CTD (15–30 %) > CHD (10–23 %) > PoPH (5–15 %)1,5
Insbesondere ältere Patientinnen und Patienten ( ≥ 65 Jahre) mit kardiovaskulären Komorbiditäten sind von der PAH betroffen.1,6 Die HPAH tritt bei jüngeren Personen auf, wobei Frauen doppelt so oft wie Männer erkranken.1
Die PAH wird abhängig von Risikofaktoren für eine Linksherzerkrankung (arterielle Hypertonie, koronare Herzkrankheit, Diabetes mellitus, Vorhofflimmern, BMI > 30 kg/m2) unterschieden.7 Bei der typischen PAH liegen weniger als drei dieser Risikofaktoren vor, bei der atypischen mehr als drei.7
Klinische Charakterisierung, Schweregrad und Prognose
Neben dem Leitsymptom Dyspnoe ist die klinische Manifestation der PAH unspezifisch.1,4,6 Dies erschwert eine schnelle Diagnose und Einleitung der Therapie – die Latenzzeit der Diagnose beträgt mehr als zwei Jahre.1,6
Die Symptome sind hauptsächlich auf die fortschreitende Rechtsherzhypertrophie sowie Dilatation des rechten Ventrikels (RV), resultierend aus der progressiven pulmonalen Vaskulopathie, zurückzuführen.1,6 Weitere klinische Anzeichen treten infolge der assoziierten Grunderkrankungen (z. B. CTD, HIV, CHD) und Komorbiditäten (z. B. kardiovaskuläre Erkrankungen) auf.1,6
Die PAH ist eine chronisch-progressive und nicht heilbare Krankheit.2,4 Insbesondere die IPAH und HPAH sind mit einer schlechten Prognose assoziiert.2,4 Die Mortalität ist aufgrund zunehmender Therapieoptionen gesunken – die Drei-Jahres-Überlebensrate der PAH liegt bei 70–80 %.3,8
Den höchsten prädiktiven Wert für die Mortalität hat die Einteilung nach WHO-Funktionsklassen (WHO-FC).1
Class | Descriptiona |
|---|---|
WHO-FC I | Patients with PH but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnoea or fatigue, chest pain, or near syncope |
WHO-FC II | Patients with PH resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnoea or fatigue, chest pain, or near syncope |
WHO-FC III | Patients with PH resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnoea or fatigue, chest pain, or near syncope |
WHO-FC IV | Patients with PH with an inability to carry out any physical activity without symptoms. These patients manifest signs of right HF. Dyspnoea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity |
Tab. 2: WHO-Funktionsklassen (WHO-FC) der pulmonalen Hypertonie (PH).a Funktionelle Klassifizierung der PH, modifiziert nach der New York Heart Association (NYHA) gemäß WHO 1998. Modifiziert nach 1.
Neben der Einteilung nach NYHA-Klassen gemäß WHO-FC werden zur Kategorisierung des Schweregrades und der einzuleitenden Therapiemaßnahmen u. a. eine klinische Bewertung, Belastungstests, biochemische Marker (BNP/NT-proBNP), Auswertung der Echokardiographie und hämodynamische Parameter herangezogen.1 Damit werden die Risikoklassen in die Stufen niedrig, intermediär und hoch eingeteilt.1
Determinants of prognosis (estimated 1-year mortality) | Low risk (<5%) | Intermediate risk (5-20%) | High risk (>20%) |
|---|---|---|---|
Clinical observations and modifiable variables | |||
Signs of right HF | Absent | Absent | Present |
Progression of symptoms and clinical manifestations | No | Slow | Rapid |
Syncope | No | Occasional syncopea | Repeated syncopeb |
WHO-FC | I, II | III | IV |
6MWDc | >440 m | 165–440 m | <165 m |
CPET | Peak VO2 >15 mL/min/kg (>65% pred.) | Peak VO2 11–15 mL/min/kg (35–65% pred.) | Peak VO2 <11 mL/min/kg (<35% pred.) |
Biomarkers: BNP or NT-proBNPd | BNP <50 ng/L NT-proBNP <300 ng/L | BNP 50–800 ng/L NT-proBNP 300–1100 ng/L | BNP >800 ng/L NT-proBNP >1100 ng/L |
Echocardiography | RA area <18 cm2 No pericardial effusion | RA area 18–26 cm2 TAPSE/sPAP 0.19–0.32 mm/ mmHg | RA area >26 cm2 Moderate or large pericardial effusion |
cMRIe | RVEF >54% RVESVI <42 mL/m2 | RVEF 37–54% SVI 26–40 mL/m2 RVESVI | RVEF <37% RVESVI >54 mL/m2 |
Haemodynamics | RAP <8 mmHg CI ≥2.5 L/min/m2 SVI >38 mL/m2 SvO2 >65% | RAP 8–14 mmHg CI 2.0–2.4 L/min/m2 SVI 31–38 mL/m2 SvO2 60–65% | RAP >14 mmHg CI <2.0 L/min/m2 SVI <31 mL/m2 SvO2 <60% |
Tab. 3: Umfassende Risikobewertung der PAH, eingeteilt in drei Risikostufen. Modifiziert nach 1.
Die Risikostratifizierung während der Nachbeobachtung basiert u. a. auf WHO-FC, Sechs-Minuten-Gehtest und biochemischen Markern (BNP/NT-proBNP).1 Die Risikoklassen werden dann unterschieden in niedrig, intermediär-niedrig, intermediär-hoch und hoch.
Determinants of prognosis | Low risk | Intermediate–low risk | Intermediate–high risk | High risk |
|---|---|---|---|---|
Points assigned | 1 | 2 | 3 | 4 |
WHO-FC | I or IIa | - | III | IV |
6MWD, m | <440 | 320–440 | 165–319 | <165 |
BNP or NT-proBNP,a ng/L | <50 <300 | 50–199 300–649 | 200–800 650–1100 | >800 >1100 |
Tab. 4: Parameter zur vereinfachten Risikobewertung der PAH mit vier Risikostufen. Modifiziert nach 1.
Diagnostik
Den diagnostischen Algorithmus beim Verdacht auf PAH finden Sie hier. Wird eine PAH vermutet, muss eine umgehende Überweisung („Fast-Track-Referral“) in ein spezialisiertes PH-Expertenzentrum für eine interdisziplinäre Betreuung erfolgen.1,2
Eine frühzeitige Diagnose ist aufgrund des progressiven Verlaufs bei allen Formen der PAH relevant.1,2
Diagnostikverfahren
Echokardiographie: Stellt das wichtigste nicht invasive Verfahren zur Diagnose und Verlaufskontrolle der PAH dar.1–3
Rechtsherzkatheter (RHC): Goldstandard für die PH-Diagnose und Klassifizierung1–4,6
Vasoreaktivitätstestung: Zur Identifizierung von Respondern auf Kalziumkanalblocker, wird nur bei Verdacht auf IPAH, HPAH und DPAH empfohlen (mit inhalativem Stickstoffmonoxid [NO] oder inhalativem Iloprost)1,2
Weitere Untersuchungen: Sechs-Minuten-Gehtest, hämatologische, immunologische sowie Biomarker-Tests (BNP/NT-pro-BNP)1
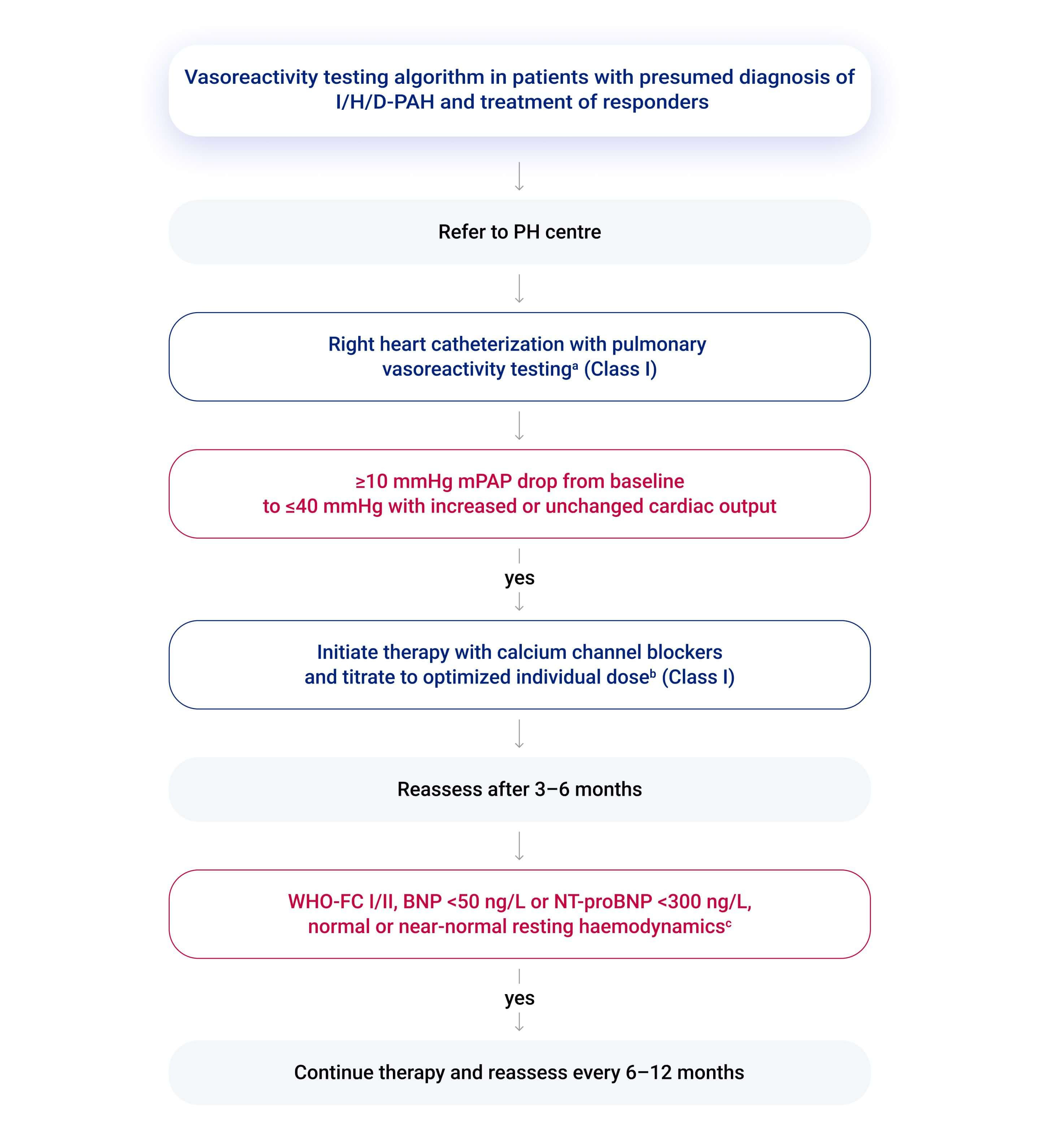
Abb. 1: Vasoreactivity testing algorithm of patients with presumed diagnosis of idiopathic, heritable, or drug-associated pulmonary arterial hypertension.
BNP, brain natriuretic peptide; I/H/D-PAH, idiopathic, heritable, drug-associated pulmonary arterial hypertension; mPAP, mean pulmonary arterial pressure; N, no; NT-proBNP, N-terminal pro-brain natriuretic peptide; PH, pulmonary hypertension; PVR, pulmonary vascular resistance; WHO-FC, World Health Organization functional class; WU, Wood units; Y, yes. aInhaled nitric oxide and inhaled iloprost are recommended; intravenous epoprostenol can be used if inhaled nitric oxide or inhaled iloprost are unavailable. bSee text for details. cmPAP ≤30 mmHg and PVR ≤4 WU.
Weiterführende Informationen zur Diagnostik finden Sie hier und eine ausführliche Beschreibung in den aktuellen ESC/ERS-Leitlinien (2022) zur PH.1
Therapie
Die Therapie der PAH ist komplex und muss in akkreditierten PH-Expertenzentren unter Berücksichtigung der Risikostratifizierung und der Begleiterkrankungen eingeleitet und überwacht werden.1,2 Therapieziel ist der Erhalt einer niedrigen Risikoklasse mittels optimierter Behandlungsstrategie und ggf. Therapieeskalatation.1
Momentan bestehen folgende zugelassene Therapieoptionen, die u.a. in Abhängigkeit des therapeutischen Algorithmus sequentiell oder in Kombination eingesetzt werden:1,2,4
Positiver Vasoreaktivitätstest (IPAH, HPAH, DPAH):
Hochdosierte Kalziumkanalblocker
PAH-Diagnose und negativer Vasoreaktivitätstest:
Endothelin-Signalweg:
Endothelin-Rezeptor-Antagonisten, ERA (p. o.) wie Bosentan, Ambrisentan, Macitentan
NO-sGC-cGMP-Signalweg:
Phophodiesterase-Typ-5-Inhibitoren, PDE5i (p. o.) wie Sildenafil, Tadalafil
Stimulatoren der löslichen Guanylatzyklase, sGC (p. o.) wie Riociguat
Prostazyklin-Signalweg:
Prostazyklin-Analoga, PCA (p. o., p. i., i. v., s. c.) wie Iloprost, Epoprostenol oder Treprostinil
Prostazyklin-Rezeptor-Agonisten, PRA (p. o.) wie Selexipag
Behandlung der zugrunde liegenden Erkrankung
Operative Versorgung bei CHD
Lungentransplantation
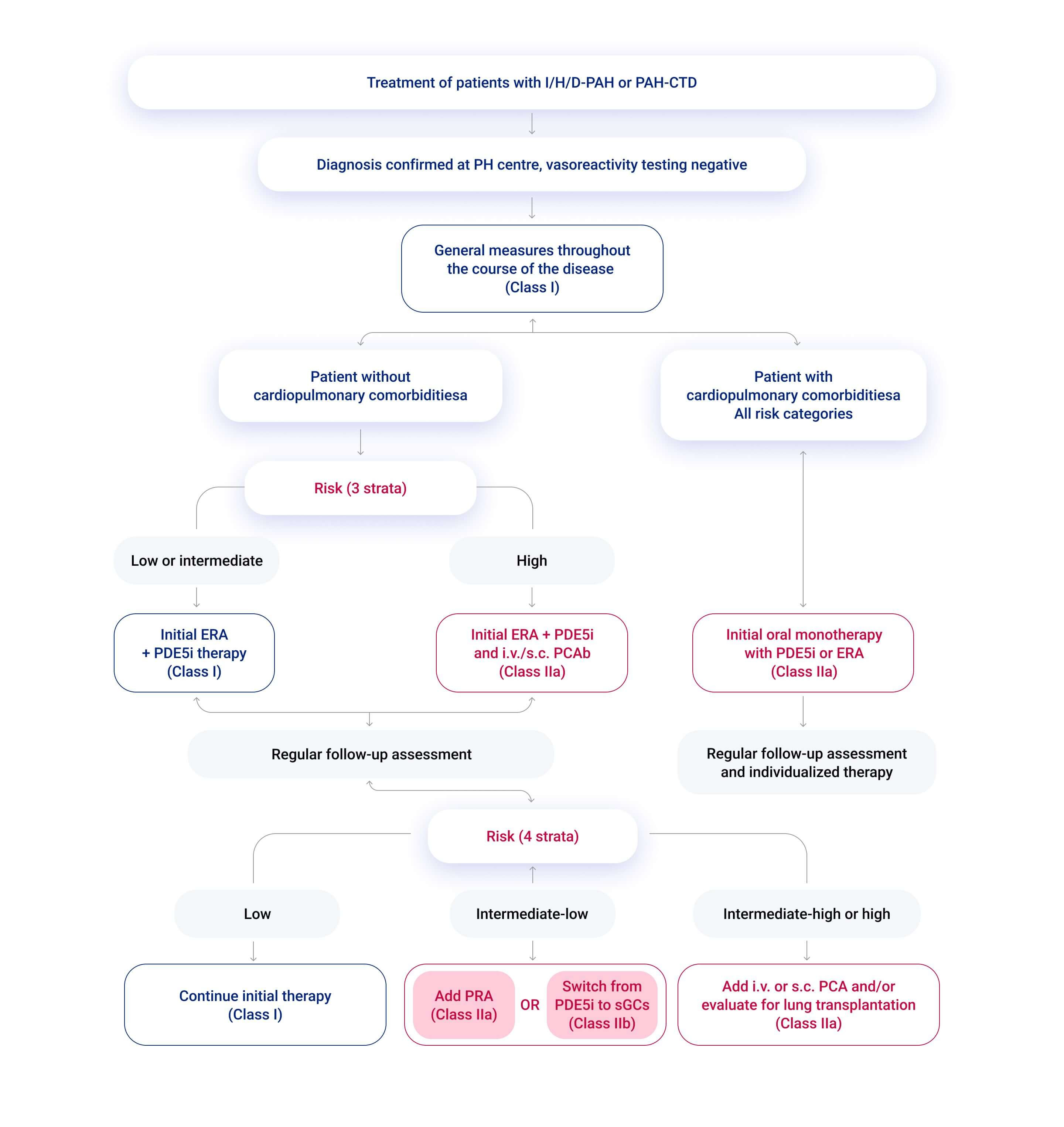
Abb. 2: Algorithmus zur Therapie von Patientinnen und Patienten mit I/H/D-PAH oder PAH-CTD. Modifiziert nach 1.
Eine ausführliche Beschreibung des therapeutischen Algorithmus ist in den aktuellen ESC/ERS-Leitlinien (2022) zur PH zu finden.1
Verdacht auf PAH?
Bei Verdacht auf PAH nutzen Sie unseren Expertenfinder und kontaktieren Sie unsere PH-Experten direkt hier auf der Plattform oder ein PH-Expertenzentrum in Ihrer Nähe.

BNP/NT-proBNP: Brain natriuretic peptide/ N-terminales pro-BNP
CHD: Angeborener Herzfehler
CPET: Cardiopulmonary Exercise Testing
cMRI: Cardiac Magnetic Resonance Imaging
CTD: Bindegewebserkrankungen
CTEPH: Chronisch-thromboembolische pulmonale Hypertonie
DPAH: Medikamenten- oder Toxin-induzierte PAH
ERS: European Respiratory Society
ESC: European Society of Cardiology
HPAH: Hereditäre PAH
IPAH; Idiopathische PAH
mPAP: Mittlerer Pulmonalarteriendruck
NYHA: New York Heart Association
PAH: Pulmonale arterielle Hypertonie
PCH: Pulmonale kapilläre Hämangiomatose
PH: Pulmonale Hypertonie
PAWP: Pulmonalarterienverschlussdruck
PoPH: Pfortaderhochdruck
PPHN: Persistierende PH des Neugeborenen
PVOD: Pulmonale veno-okklusive Erkrankung
RHC: Rechtsherzkatheteruntersuchung
RV: Rechter Ventrikel
WHO-FC: Funktionsklasse eingeteilt durch die World Health Organziation
1. Humbert M et al. Eur Heart J 2022; 43(38): 3818–3731.
2. DGPK, S2k-Leitlinie Pulmonale Hypertonie, Version 3.0, Stand 29.04.2020, verfügbar unter: https://register.awmf.org/de/leitlinien/detail/023-038 (letzter Zugriff am 08.04.2024).
3. Foris V et al. Pneumologe 2021; 18: 195–206.
4. Rosenkranz S et al. Herz 2023; 48(1): 23–30.
5. Lau EMT et al. Nat Rev Cardiol 2017; 14(10): 603–614. Epub 2017 Jun 8.
6. Maron BA et al. Am J Respir Crit Care Med 2021; 203(12): 1472–1487.
7. Opitz CF et al. J Am Coll Cardiol 2016; 68(4): 368–378.
8. Hoeper MM et al. Int J Cardiol 2013; 168(2): 871–80.